

Эффективность лечения и профилактики заболеваний человека во многом зависит от применения качественных лекарственных препаратов (ЛП). Мировая медицинская практика показывает, что использование недостаточно исследованных, некачественных или контрафактных препаратов может вызывать серьезные побочные действия и приводить к нежелательным реакциям, вплоть до гибели пациентов. Примерами этому могут служить вспышки синдрома эозинофильной миалгии в 1989 г., в результате приема генно-инженерного L-триптофана (Showa Denko), что привело к гибели 37 пациентов; эпидемия грибкового менингита в США в 2012 г., вызвавшая заболевание более 750 человек и гибель 64, в результате применения загрязненного спорами грибка Exserohilum rostrum препарата метилпреднизолона ацетата (New England Compounding Center), гибель пациентов в Китае, в результате применения вакцины от гепатита В (BioKangtai) в 2013 г., другие подобные случаи [9]. В России, в последние годы, согласно данным официальной статистики, количество некачественных препаратов держится на высоком уровне [3]. Поэтому обеспечение населения безопасными, эффективными, качественными и конкурентоспособными ЛП является основной задачей стратегии развития отечественной фармацевтической промышленности. Определяющим критерием качества ЛП при выпуске на производстве является его соответствие требованиям нормативной документации (НД), гарантирующее подлинность, чистоту, эффективность и безопасность применения в медицинской практике ЛП. Однако, следует отметить, что различия в стандартах качества фармацевтического производства в разных странах мира приводят к появлению на отечественном фармацевтическом рынке аналогичных ЛП различного качества.
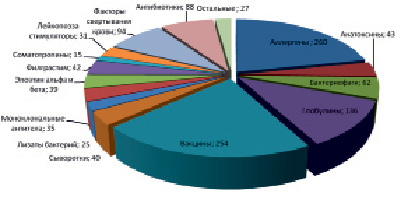
Распределение основных групп БЛП, зарегистрированных в РФ
Современный этап развития фармацевтической индустрии характеризуется разработкой высокотехнологичных наукоемких производств лекарственных средств биологического происхождения (БЛС), действующее вещество которых произведено или выделено из биологического источника и для определения свойств и качества, которых необходима комбинация биологических и физико-химических методов, к которым согласно Федеральному закону № 61-ФЗ «Об обращении лекарственных средств» следует относить:
– иммунобиологические лекарственные препараты (вакцины, анатоксины, токсины, сыворотки, иммуноглобулины и аллергены и др.);
– биотехнологические лекарственные препараты (БтЛП) (в том числе полученные методами ДНК-рекомбинантной технологии, технологии контролируемой экспрессии генов, кодирующих биологически активные белки в прокариотах и эукариотах, включая измененные клетки млекопитающих, гибридомного метода и метода моноклональных антител);
– лекарственные препараты, полученные из крови, плазмы крови человека и животных (за исключением цельной крови);
– генотерапевтические лекарственные препараты.
Технология получения БЛС – сложный многостадийный процесс, некоторые этапы которого, такие как: получение субстанции, готового продукта, фасовка в первичную и упаковка во вторичную (потребительскую) упаковку – чаще всего осуществляют не на одной производственной площадке, а на разных (в одной стране или нескольких государствах). В результате возникают трудности при стандартизации производства, увеличивается вероятность отклонений от исходного валидированного процесса, что может влиять на качество готового продукта. При этом, даже незначительные изменения технологии способны привести к снижению эффективности и/или увеличению токсичности, и, в конечном счете, могут стать причиной неэффективности терапии и возникновение нежелательных явлений у пациентов. Статистика по БтЛП препаратам, показывает, что более 10 % серьёзных побочных эффектов новых лекарств невозможно выявить, несмотря на тщательно проводимые исследования на безвредность [3]. Поэтому обеспечению и оценке безопасности новых классов биологических ЛП в процессе производства должно уделяться повышенное внимание.
Одной из основных причин увеличения токсичности ЛП может являться образование в их составе неожиданных токсических примесей (НТП) в процессе производства, хранения, или транспортировки с нарушением установленных норм. Сложность обнаружения НТП заключается в их непредсказуемости, что не позволяет выявить такие примеси в рутинном посерийном контроле качества ЛС. Физико-химические методы (такие как ВЭЖХ, спектроскопия, и др.), в связи с высокой специфичностью к искомому аналиту и ограниченным набором методов и показателей качества, закрепленных в НД, как правило, не позволяют обнаруживать подобные примеси. Иными словами, необходимо знать искомую примесь, так как для ее определения требуется разработка методики, с выбором индивидуальных условий анализа для включения в НД.
На сегодняшний день для выявления НТП используется биологический тест – «Аномальная токсичность». Он включен во все ведущие фармакопеи мира (USP – «Safety tests», Eur.Ph – «Abnormal toxicity», Br.Ph. – «Abnormal toxicity», страны ЕврАзЭс – «Аномальная токсичность»). Однако нельзя проигнорировать тот факт, что ведется активная дискуссия о том, следует ли включать данный показатель качества в НД или возможно не оценивать качество ЛП по данному показателю. Целым рядом фармпроизводителей активно распространяется мнение о необходимости исключения показателя «Аномальная токсичность» из НД на ЛП. Данной позиции придерживается и Европейская федерация фармацевтических производителей EFPIA [1,8]. Основными доводами в пользу исключения теста на животных являются такие аргументы как: соблюдение производителями правил GMP, использование валидированных процессов производства, неспецифичность метода «Аномальная токсичность», наличие методов подтверждения отсутствия загрязняющих веществ различного типа (бионагрузка, микробиологическая обсемененность, наличие бактериальных эндотоксинов и других пирогенов), проведение посерийного выпускающего контроля качества, отсутствие требований EMA и FDA по проведению теста на аномальную токсичность при контроле качества готовой продукции, принятие директивы по защите животных, используемых в научных целях (Directive 2010/63/EU of 22 September 2010). В качестве альтернативы тесту «Аномальная токсичность» предлагается использовать физико-химические методы, а также биологические методы: «Бактериальные эндотоксины», «Пирогенность», «Микробиологическая чистота» и тест на стерильность.
В то же время большинство российских исследователей придерживаются иного мнения, указывая на то, что замена теста «Аномальная токсичность» физико-химическими методами анализа преждевременна и возможна только после проведения масштабных сравнительных исследований, которые не всегда оканчиваются положительными результатами в пользу аналитических методов. Убедительные данные о недостаточности использования только физико-химических методов при выявлении НТП получены даже для хорошо стандартизированных производств синтетических препаратов, возможность же подобных случаев для ЛП биологического происхождения многократно возрастает. Так сравнительный анализ результатов испытания «Аномальная токсичность» и содержания примесей в субстанции дротаверина гидрохлорида в различных модификациях метода ВЭЖХ не позволил авторам выбрать приемлемую методику контроля родственных примесей [2]. Было установлено, что все методики, предлагаемые для контроля родственных примесей в субстанции дротаверина гидрохлорида дают отличные друг от друга значения, как общего содержания примесей, так и их числа, и, следовательно, не позволяют рекомендовать ни одну из них как правильную для включения в фармакопею с целью контроля качества вышеуказанной субстанции. При этом испытание «Аномальная токсичность», выполненное в соответствии с требованиями Европейской Фармакопеи, установило, что все исследуемые образцы препарата формально выдерживают испытание по количественному учету выживаемости мышей, но при этом вызывают различные по выраженности клинические явления интоксикации, что доказало наличие в субстанции дротаверина гидрохлорида высокотоксичных примесей, которые следует идентифицировать, регламентировать, и контролировать их содержание. Подобные примеры встречаются и при испытании БЛП, например для препаратов холина альфосцерата, антибиотиков цефалоспоринового ряда, фосфолипидных препаратов и др., что подтверждает наличие в них НТП не определяемых аналитическими методами.
Интересные результаты были получены при изучении НТП в БтЛП. Их белковая природа способствует образованию целого ряда примесей, которые возможно детектировать только на биологических тест-объектах, и невозможно определить физико-химическими методами. В последние годы, появились данные о возникновении различных негативных реакций на подобные препараты, имеющие в составе в качестве стабилизаторов полисорбаты, которые по мнению исследователей образуют пероксидные производные и другие примеси, что вызывает данные осложнения. Согласно литературным данным [6, 7] в некоторых случаях помимо структурных комплексов Полисорбата-80 и белка образуются трудно определяемые частицы (остаточные свободные химические соединения), способные на реакцию с белками, что может привести к повышению иммуногенности. В случае, если полисорбат находится в водном растворе, ускоряется процесс самоокисления, а количество частиц, вызывающих реакцию, постоянно увеличивается как в течение всего процесса производства, так и во время хранения, вплоть до момента применения лекарственного препарата.
Aditya A. Wakankar с соавторами (2010) [10] показали, что экстрагируемые и вымываемые вещества из первичной упаковки являются примесями, которые возникают в результате контакта препарата с такими компонентами, как прокладки, пробки, картриджи и предварительно заполненные шприцы, которые используются для обработки, хранения и/или доставки биопрепаратов. Эти примеси могут потенциально оказывать влияние на качество и безопасность лекарственного препарата, непосредственно или косвенно воздействовать на пациента путем взаимодействия с белками, образуя белковые аддукты. Поэтому при разработке биологических препаратов с различной композицией вспомогательных компонентов необходимо учитывать все экзо- и эндогенные факторы, исследовать профили деградации белков и совместимость препарата с различными материалами/поверхностями первичной упаковки. Одним из подходов для изучения, которых, может быть выполнение параллельных исследований стабильности образцов БтЛП в стрессовых условиях по показателям «Аномальная токсичность» и «Чистота. Посторонние примеси», что возможно позволит получить доказательства отсутствия или, наоборот, негативного воздействия на организм появившихся при деградации активного компонента примесей.
Разнообразный спектр НТП, способных образовываться в составе БЛП, требует применения универсального метода для их выявления вне зависимости от механизма токсического действия последних на живой организм. И именно тест «Аномальная токсичность», на сегодняшний день, наиболее полно отвечает данному требованию. Однако, важно понимать, что только правильное применение данного показателя, позволяет снизить риск поступления на фармацевтический рынок небезопасных ЛП, а нарушение методических основ испытания приводит к невоспроизводимости метода, в других случаях – бессмысленности его проведения.
Одной из наиболее важных причин, которая дискредитирует надежность испытания «Аномальная токсичность», являются неадекватно подобранные тест-доза, скорость введения и другие условия испытания. Для эффективного контроля безопасности ЛП тест «Аномальная токсичность» должен выполняться в оптимальных и достаточно жестких условиях: необходимо использовать, как правило, внутривенный путь введения и тест-дозу, близкую к предельно переносимой, которая дает возможность выявить превышение установленного ранее уровня токсичности препарата, что является маркером изменений в составе препарата при накоплении токсических примесей. Зачастую недобросовестные производители, умышленным занижением требований к условиям тестирования препарата компенсируют его качество. Подобные примеры встречаются достаточно часто, для разных групп биологических ЛП. Так например, для препаратов группы антибиотиков у ряда производителей в проекте НД на стрептомицин предлагается снизить тест-дозу с общепринятой 1,3 мг/мышь до 1,0 мг/мышь. Для препаратов гентамицина в ряде проектов НД предлагается увеличить время введения препарата до 15-30 секунд, что приводит соответственно к уменьшению скорости введения 0,1 мл/3-6 с. Наиболее демонстративным является случай с препаратом цефалоспоринового ряда «Цефтриаксон, порошок для приготовления раствор для внутривенного и внутримышечного введения, 1 г», где, в нормативной документации одной из фирм для контроля качества по показателю «Аномальная токсичность» были предложены следующие условия испытания: тест-доза 30 мг в 0,5 мл раствора на мышь, при введении за 60 секунд. Данные условия значительно «мягче» фармакопейной прописи ГФ РФ, где регламентирована скорость введения 0,1 мл/сек (т.е. время введения 5 секунд). При экспертизе качества трех серий данного ЛП, которые были изготовлены из субстанций трех различных производителей, согласно требованиям НД, все серии соответствовали спецификации. При постановке теста в соответствии с требованиями ГФ РФ в общепринятых условиях, была выявлена аномальная токсичность одной из серий препарата. Это показывает бoльшую информативность испытания, выполненного в адекватных условиях, по сравнению с тестом, предложенным в НД. Важно отметить, что использование в данном случае физико-химических аналитических методов (согласно НД), не позволили выявить различия в качестве анализируемых серий.
Большое значение в снижении воспроизводимости результатов разных производителей имеют и существующие на сегодняшний день различия в методических подходах к тесту «Аномальная токсичность» в ведущих фармакопеях мира [1, 4]. Прежде всего, выявлены основные различия в условия проведения Российской и Европейской фармакопеями – значительный разброс по массе используемых животных (ЕР 17,0-24,0 г; ГФ 19,0-21,0 г) и в разной скорости введения испытуемого препарата 0,1 мл/с (ГФ РФ) и 0,3 мл – 0,6 мл/с (ЕФ). Так, уменьшение времени введения испытуемого препарата с 5 с до 30 с маскирует истинные реакции интоксикации на неспецифические примеси, уменьшая их выраженность [5], и приводит к необходимости пересмотра утвержденных тест-доз, в противном случае делает данное испытание неинформативным.
Немало проблем возникает при включении показателя «Аномальная токсичность» в НД на препараты моноклональных антител, что связано со сложностью классификации данных ЛП, их видовой специфичностью и некоторыми другими аспектами. Одной из наиболее распространенных ошибок, встречающихся в НД на препараты моноклональных антител (МАБ) является постановка испытания «Аномальная токсичность» в варианте «Тест для иммунобиологических лекарственных препаратов». Препараты данной группы являются антителами, полученными с применением технологий моноклональных антител, поэтому, согласно 61-ФЗ, относятся к группе биотехнологических ЛП, т.е. «Аномальная токсичность» должна оцениваться в соответствии с основным разделом и выполняться только на пяти мышах, при внутривенном введении со скоростью 0,1 мл/сек, в тест-дозе близкой к максимально переносимой. При этом отсутствие терапевтической релевантности гуманизированных антител у мышей не является препятствием к оценке токсичности МАБ на данном виде животных, поскольку на сегодняшний день отсутствуют убедительные доказательства о вкладе механизма терапевтического эффекта в реализацию токсического действия, которое проявляется, преимущественно, за счет вспомогательных компонентов, входящих в состав МАБ.
Следует отметить, что, как и стандартные физико-химические методы, биологические методы («Пирогенность», «Бактериальные эндотоксины» «Стерильность», «Микробиологическая чистота»), перечисленные в обзорах EFPIA и Й.Х.Гарбе, также не позволяют выявить НТП в составе биологических ЛП. Такие показатели как «Стерильность» и «Микробиологическая чистота» направлены на выявление жизнеспособных микроорганизмов в испытуемых препаратах, а показатели «Пирогенность» и «Бактериальные эндотоксины» – устанавливают наличие пирогенных примесей, и в первую очередь разрушенных бактериальных клеток [4]. Поэтому даже включение данных показателей в НД не может гарантировать отсутствие аномальной токсичности ЛП, за счет образования НТП. Предлагаемые в рамках принятия Директивы 2010/63/EU и концепции 3R, испытания в опытах in vitro с использованием культур клеток, субклеточных структур или других подобных тест-объектов также не способны заменить эксперименты на целостном организме и могут рассматриваться только как дополнительные исследования. Такие методики, как правило, не стандартизованы и даже при использовании одних и тех же культур клеток, значительно различаются между собой. Кроме того для выявления максимального спектра НТП необходимо использовать не одну модель, а несколько тестов, что значительно увеличивает стоимость и трудоемкость контроля. Таким образом, на сегодняшний день не разработаны абсолютно надежные методы проверки безвредности БЛП, альтернативные показателю «Аномальная токсичность». В свете этого доводы, приводимые в пользу исключения данного испытания, не являются убедительными и доказательными. В то же время, опыт работы при проведении контроля качества ЛП показывает, что на фоне тенденции к исключению испытания «Аномальная токсичность» из НД качество ЛП не улучшается. Только за I квартал 2016 года число выявленных несоответствий по данному показателю составило 5,4 % в то время как в предыдущие 10 лет доля данного показателя не превышала 2,5 % [3]. Учитывая необходимость, со стороны регулирующих органов, обеспечить безопасность фармацевтической продукции, для всех регистрируемых в РФ лекарственных препаратов, вне зависимости от степени внедрения GMP, должны предъявляться единые требования к спецификациям выпускаемой продукции. Поэтому перед выходом на Российский рынок серии БЛП, выпущенные зарубежными производителями, должны пройти испытания на аномальную токсичность, что является общеприменимой мировой практикой при отсутствии межгосударственных договоров о взаимном признании результатов испытаний и уверенности в безопасности биологических препаратов.
В соответствии с требованиями, предъявляемыми в РФ, показатель «Аномальная токсичность» является обязательным для всех биологических ЛП, преимущественно для парентерального применения. Данное требование включено в Государственную фармакопею Российской Федерации ХIII издания в ОФС 1.2.4.0004.15 «Аномальная токсичность», ОФС 1.1.0006.15 «Фармацевтические субстанции», «Лекарственные средства, получаемые методами рекомбинантных ДНК», ОФС 1.8.1.0002.15, ОФС 1.4.1.0007.15 «Лекарственные формы для парентерального применения», ОФС 1.7.1.0007.15 «Иммунобиологические препараты», ОФС 1.7.1.0003.15 «Вакцины и анатоксины», ОФС 1.7.1.0001.15. «Аллергены», ОФС 1.7.1.0002.15 «Бактериофаги», ОФС 1.8.1.0003.15 «Иммуноглобулины человека» и ОФС 1.8.1.0004.15 «Иммуноглобулины и сыворотки (антитела) гетерологичные».
В то же время, общая глобализация и унификация требований меняют в настоящее время устоявшиеся подходы к системе оценки качества ЛС. Заметно интенсифицировался процесс интеграции стран-участников ЕврАзЭс, в частности в отношении подготовки единой нормативно-правовой базы в системе обращения лекарственных средств внутри Союза. Рекомендации международных сообществ оказывают значительное влияние на развитие новых и пересмотр существующих норм и правил.
Заключение
Объем мирового рынка биотехнологий на сегодняшний день оценивается в 270 млрд долларов, а прогнозируемые темпы роста составляют 10-12 % в год до 2020 года. Ожидается его увеличение до 600 млрд долларов к 2020 г. Проведение испытания на аномальную токсичность является одним из гарантов безопасности биотехнологических препаратов, а дискуссии по исключению данного теста из нормативной документации носят голословный характер. Отсутствуют фактические материалы и данные, доказывающие безопасность спонтанных примесей, появление которых возможно при нарушениях условий хранения, транспортирования, изменениях, вносимых в технологию производства. Принимая во внимание рекомендации ВОЗ производитель должен представить на рассмотрение информацию «…с объяснением, что либо способ введения препарата, либо методы его изготовления, либо природа самого продукта являются доказательством, что тест общей токсичности не нужен для подтверждения безопасности, чистоты и активности продукта». Такие же доказательства нужно представить в случае невозможности выполнения теста общей токсичности в силу каких-либо обстоятельств. Если это возможно, следует предложить альтернативные процедуры для подтверждения безопасности препарата.
Таким образом, рассматривать возможность исключения показателя «Аномальная токсичность» следует только при убедительных экспериментальных доказательствах.
Библиографическая ссылка
Рябцева М.С., Филимонова И.Н., Осипова И.Г., Неугодова Н.П., Ковалева Е.Л., Шаройкина М.В. ПОКАЗАТЕЛЬ КАЧЕСТВА «АНОМАЛЬНАЯ ТОКСИЧНОСТЬ» – ОДНА ИЗ ОСНОВНЫХ СОСТАВЛЯЮЩИХ БЕЗОПАСНОСТИ БИОЛОГИЧЕСКИХ ЛЕКАРСТВЕННЫХ СРЕДСТВ // Международный журнал прикладных и фундаментальных исследований. – 2016. – № 9-2. – С. 232-237;URL: https://applied-research.ru/ru/article/view?id=10225 (дата обращения: 24.04.2024).