

Среди обширного спектра хромосомных и геномных аномалий, обнаруживаемых у детей с задержкой психоречевого и психомоторного развития, врождёнными пороками и микроаномалиями развития, а также различными неврологическими нарушениями, значительное место занимают дупликации и трипликации участков отдельных хромосом [1–3]. Размеры дуплицированных участков сильно варьируют, от нескольких сот пар нуклеотидов до десятков миллионов пар; клинические проявления при этом зависят не только от размеров дупликации, но и от генов, находящихся в дуплицированном участке, и могут быть как очень тяжёлыми, так и средней тяжести [4]. Некоторые часто встречающиеся микродупликации с повторяющимися клиническими признаками описывают как микродупликационные синдромы. Как правило, дупликации и трипликации регулярны, но встречаются и мозаичные случаи. Диагностика таких случаев нередко требует применения таких молекулярно-цитогенетических методов, как флюоресцентная гибридизация in situ (FISH) с различными ДНК зондами, многоцветное окрашивание хромосом (МСВ), метафазная геномная гибридизация (HR CGH) и серийная сравнительная геномная гибридизация (молекулярное кариотипирование) на ДНК-микроматрицах (arrayCGH), что позволяет выявлять делеции и дупликации, определять точки разрыва при них и размер дуплицированного участка [5–7]. Имея точные координаты перестройки, можно анализировать гены, находящиеся в дуплицированном участке, для проведения корректного медико-генетического консультирования семьи с поражённым ребёнком [8, 9]. В работе представлен случай дупликации длинного плеча хромосомы 15 у ребёнка с врождённым пороком сердца, сниженным интеллектом, энурезом и энкопрезом.
Материалы и методы исследования
В работе обследован ребёнок женского пола 6 лет. Культура лимфоцитов периферической крови, приготовление препаратов, дифференциальное окрашивание хромосом по длине и анализ кариотипа проводились по стандартным методикам (GTG- и CBG-окрашивание хромосом). Серийная сравнительная геномная гибридизация (молекулярное кариотипирование или arrayCGH) на микрочипах проводилась по стандартным протоколам с применением SNP/олигонуклеотидной микроматрицы AffymetrixCytoscan HD, содержащей 2 696 550 проб. Биоинформатический анализ данных, полученных в результате молекулярного кариотипирования, проводился с помощью баз данных NCBI, OMIM, BioGps [10].
Результаты исследования и их обсуждение
В клинику института поступила девочка 6 лет, которая родилась в 1-й беременности с массой тела 3160 г, длиной – 49 см от неродственных родителей. Причиной обращения послужили следующие клинические проявления: умеренная задержка физического развития, задержка психоречевого развития, нарушение поведения, энурез и энкопрез днём, смешанный астигматизм. Ранее отмечены недифференцированная дисплазия соединительной ткани, открытый артериальный проток, пролапс митрального клапана, вальгусная деформация стоп. Неврологом обнаружены дислалия и неврозоподобный синдром с нарушениями эмоционально-волевой сферы, нарушение работоспособности и повышенная истощаемость на фоне незрелой эмоционально-волевой сферы. У ребёнка при обследовании в нашем институте наблюдали следующие микроаномалии развития: эпикант, бороздка в середине фильтра, короткие глазные щели, высокая граница роста волос на лбу, гипертелоризм сосков, сандалевидная щель, вздёрнутый нос, крыловидные лопатки. Проведена МРТ головного мозга, которая не выявила значительных отклонений. УЗИ органов брюшной полости и малого таза выявило изгиб жёлчного пузыря и аномальную его форму.
На основании клинических признаков ребёнку провели цитогенетическое исследование. В результате цитогенетического исследования (GTG- и CBG-окрашивания хромосом по длине) кариотип пробанда – 46,XX,?dup(15)(q11.?2q1?5) (рис. 1). Для верификации дупликации и уточнения точек разрыва пробанду было рекомендовано проведение молекулярно-цитогенетического исследования – серийной сравнительной геномной гибридизации (микроматричного анализа/arrayCGH). Результаты молекулярного кариотипирования следующие: arr 3q29 (195 776 591 – 195 777 111)x4, 4q26 (114 212 421-114 219 002)x1, 15q12q15.3 (26 616 861 – 43 845 410)x3.
На рис. 2 показаны результаты проведённого ребёнку молекулярно-цитогенетического исследования – серийной сравнительной геномной гибридизации (arrayCGH) с последующей приоритизацией генов и геномных сетей. В результате проведённого анализа дупликация участка длинного плеча хромосомы 15 была подтверждена. Размер дуплицированного участка – 17 228 549 пн (рис. 2). Дупликация затронула 156 генов, 82 из которых индексированы в OMIM. Кроме того, обнаружены интрагенные вариации числа копий последовательностей ДНК (рис. 2): трипликация последовательности ДНК гена TFRC (геномная локализация – 3q29; 195 776 591 – 195 777 111; размер 520 пн) и делеция последовательности ДНК гена ANK2 (геномная локализация – 4q26; 114 212 421 – 114 219 002; размер 651 пн).
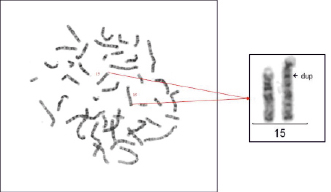
Рис. 1. Кариотип пробанда – 46,XX,?dup(15)(q11.?2q1?5). На рисунке слева метафазная пластинка лимфоцитов периферической крови с GTG-окрашиванием. Справа представлены отдельно гомологи хромосомы 15, левый из них – нормальный, правый – с дупликацией
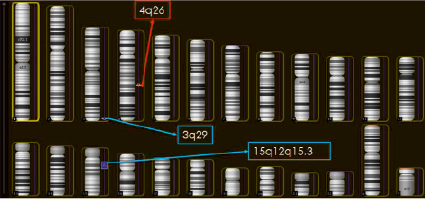
Рис. 2. Результаты молекулярного кариотипирования: CNV, обнаруженные у пробанда методом arrayCGH: трипликация 3q29 (520 пн), делеция 4q26 (651 пн), дупликация 15q12q15.3 (17 228 549 пн)
Биоинформатический (in silico) анализ
В дальнейшем был проведён биоинформатический анализ in silico для приоритизации генов, т.е. для установления корреляции обнаруженных хромосомных и геномных перестроек и фенотипа пробанда. При этом использовались базы данных NCBI, OMIM, BioGps. В области дупликации хромосомы 15 локализовано 156 генов согласно UCSC Genome Browser (GRCh37/hg19) (рис. 3). Для дальнейшего анализа влияния отдельных генов, локализованных в области дупликации, на фенотипические проявления, обнаруженные у пробанда, мы обратились к литературным источникам [11–13], провели анализ схожих случаев [14–16] и биоинформатический анализ in silico, направленный на приоритизацию генов и процессов-кандидатов.
Биоинформатический анализ участка дупликации 15q с приоритизацией генов, затронутых выявленными у пробанда хромосомными аномалиями, позволил выделить 82 гена, возможно, ассоциированных с патологическими проявлениями у пробанда.
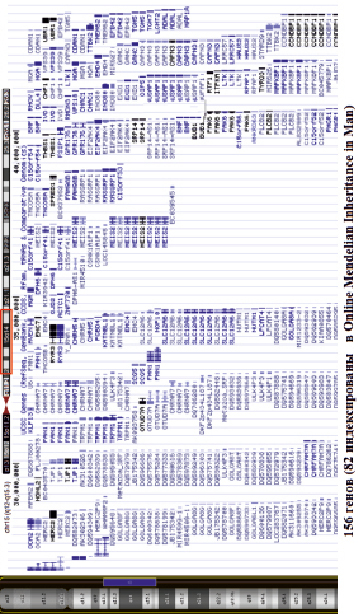
Рис. 3. Схематическое изображение дупликации хромосомы 15, обнаруженной с помощью молекулярного кариотипирования, со списком генов, локализованных в этом участке
АНАЛИЗИРУЕМЫЕ ГЕНЫ
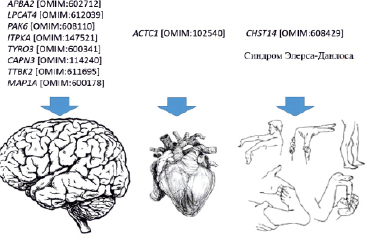
Рис. 4. Гены, расположенные в участках, затронутых аномалиями хромосом, и выделенные после приоритизации (стрелками указаны поражённые органы, ассоциированные с этими генами)
По данным литературы, ранее не было описано подобной дупликации в участке с такими точками разрыва. В ходе анализа литературы были определены гены, нарушения в которых могут быть связаны с фенотипическими проявлениями у обследованного ребёнка. Один из дуплицированных генов, ген CHST14, ассоциирован с синдромом Элерса – Данлоса. У представленного ребёнка обнаружена недифференцированная дисплазия соединительной ткани, гипермобильность суставов, пролапс митрального клапана и вальгусная деформация стоп, и эти симптомы наблюдаются при данном синдроме (рис. 4) [17, 18].
Нарушения в гене ACTC1 [OMIM:102540], по данным литературы, могут быть связаны с дефектом межпредсердной перегородки II типа и незаращением артериального протока, что и отмечено у пробанда [19, 20] (рис. 4). Среди 156 генов, согласно данным BioGps (GeneAtlas U133A), повышенная экспрессия в клетках головного мозга отмечена у 8 генов: APBA2 [OMIM:602712], LPCAT4 [OMIM:612039], PAK6 [OMIM:608110], ITPKA [OMIM:147521], TYRO3 [OMIM:600341], CAPN3 [OMIM:114240], TTBK2 [OMIM:611695], MAP1A [OMIM:600178] (рис. 4).
На рис. 4 указаны основные гены, возможно ассоциированные с фенотипическими проявлениями у пробанда. В левой части схемы – гены, связанные с нарушениями психики; в средней части – ген, при аномалиях которого возможны нарушения сердечно-сосудистой системы; справа – ген, влияющий на формирование соединительнотканных аномалий и синдрома Элерса – Данлоса.
Кумулятивный эффект изменения числа копий последовательностей ДНК данных генов-кандидатов с повышенным уровнем экспрессии в клетках головного мозга может негативно сказываться на развитии и функционировании головного мозга и всей центральной нервной системы, приводя, таким образом, к развитию патологических проявлений, описанных у пробанда.
В достаточно широком спектре различных численных и структурных аномалий хромосом значительное место занимают дупликации и трипликации хромосом. Клинические признаки при дупликациях и трипликациях сильно варьируют и зависят прежде всего от того, в какой хромосоме произошла перестройка и какие гены этой перестройкой затронуты. В целом при сравнении с другими видами хромосомных аномалий (делеции, несбалансированные транслокации, кольцевые хромосомы и др.) чаще всего патологические проявления при дупликациях бывают умеренной тяжести. Данный случай необычен тем, что при относительно больших размерах дупликации (более 17 млн пн) тяжесть клинических признаков не слишком велика, а также своеобразием симптомокомплекса (парциальный дефект психики, врождённый порок сердца, энурез и энкопрез днём, соединительнотканные нарушения). Результаты проведённых цитогенетических и молекулярно-цитогенетических исследований у данного пациента позволили корректно провести генетическое консультирование этой семьи и дать прогноз развития ребёнка, который после проведения соответствующего комплексного лечения практически не испытывает проблем с произвольным регулированием естественных потребностей, посещает общеобразовательную школу.
Заключение
Крупные геномные аномалии обнаруживаются у 25 % детей с умственной отсталостью и/или аутизмом. Обычно такие перестройки выявляют классическим кариотипированием, которое является «золотым стандартом» в цитогенетике, но этот метод анализа генома не удовлетворяет запросы генетической диагностики, так как цитогенетическая технология ограничена разрешающей способностью, а дефекты генома могут быть невелики размером. Поэтому на нынешнем уровне развития генетической диагностики необходимо использование современных молекулярно-цитогенетических методов. Кроме того, внедрение современных высокотехнологичных методов исследований (arrayCGH, FISH и др.) позволяет изучать геном с гораздо более высоким уровнем разрешения, что даёт исследователям возможность находить дополнительно те перестройки, которые, несмотря на их малый размер, могут быть основным фактором различных патологических состояний, в том числе нарушения психики. Важно отметить, что молекулярно-цитогенетические методы, особенно такие, как arrayCGH, дают исследователю огромные объёмы данных, требующих интерпретации, что невозможно без биоинформатического анализа in silico с привлечением специфических баз данных для приоритизации обнаруженных генов и генных (геномных) сетей, что позволяет определить каскады процессов, приводящих к патологическим проявлениям, и, в отдельных случаях, предложить возможную персонализированную терапию на основе знаний о механизме возникновения тех или иных нарушений. Персонифицированная терапия достоверно увеличивает качество и продолжительность жизни пациентов с различными генетическими аномалиями, а также качество жизни семей с такими детьми. Кроме того, применение современных методов генетической диагностики с привлечением специфических биоинформатических подходов к анализу генома позволит корректно проводить медико-генетическое консультирование. Результаты подобных исследований позволяют понять процессы взаимодействия генов в геномных кластерах и сетях.
Работа поддержана грантами Российского фонда фундаментальных исследований (проекты № 17-04-01366А и №16-54-76016 ЭРА_а) и программой фундаментальных исследований Президиума РАН «Фундаментальные исследования для разработки биологических технологий» (ФИМТ) (проекта № ФИМТ-2014-235).