

Интерес к исследованию соединений серии 115 на базе f-элементов вызван интересными физическими свойствами соединений данной группы. Cоединение PuCoGa5, базовое соединение серии, является сверхпроводником с высокой для соединений семейства Pu температурой сверхпроводящего перехода около 18,5 K [1]. Это значение на порядок превосходит критические температуры соответствующих изоструктурных аналогов на основе церия [2]. Очевидно, что выдающиеся физические свойства PuCoGa5 в значительной степени определяются его электронной структурой. Ранее нами для соединений серии данной серии изучена электронная структура в рамках зонных методов с учетом электронных корреляций и спин-орбитальной связи [3], предложен метод оценки оптимального уровня электронного легирования, приводящего к появлению сверхпроводящего состояния [4]. Для слоистой системы Pu(Co, Fe, Ni, Rh, Ir)Ga5 понимание физических свойств представляет большую физическую проблему [5, 6]. Несмотря на проведенные экспериментальные исследования, данных для описания поведения свойств PuNiGa5 под давлением пока в научной литературе не представлено [7]. Получение новых данных о физических свойствах, электронной структуре, магнитных и спектральных характеристиках соединений данной серии, а также понимание роли переходных металлов в этих слоистых соединениях позволят совершенствовать технологии работы с данными материалами и в перспективе существенно расширить спектр их применения.
В этой статье мы приводим результаты первопринципных расчетов электронной структуры интерметаллида PuNiGa5 с целью получения более глубокого понимания поведения его электронной структуры и магнитных свойств под давлением, а также исследуем влияние электронных корреляций на электронную структуру интерметаллида PuNiGa5 при нормальных условиях.
Материалы и методы исследования
Соединение PuNiGa5 кристаллизуется в тетрагональной структуре c пространственной группой симметрии Р4/mmm (номер группы 123). Элементарная ячейка содержит одну формульную единицу с атомом Pu в кристаллографической позиции 1а (0, 0, 0), атом никеля располагается в позиции типа (0, 0, 1/2) и 5 атомов Ga – в позициях типа (1/2, 1/2, 0) и (0, 1/2, 0,307). В расчетах использовались экспериментальные величины параметров кристаллической решетки, указанные в работе [7].
Расчеты электронной структуры выполнены в приближении локальной электронной плотности с поправкой на кулоновские корреляции и спин-орбитальную связь (LDA + U + SO) [3, 4]. Использовался пакет программ TB-LMTO-ASA на основе метода линеаризованных маффин-тин орбиталей в приближении атомных сфер. В литературе на сегодняшний день можно найти несколько тысяч публикаций, основанных на результатах исследований при помощи метода LDA + U. Данный метод доказал свою исключительную полезность при описании физических свойств многих классов материалов, включая соединения с дальним магнитным порядком. Интегрирование методом тетраэдров осуществлялось по сетке k-точек в обратном пространстве. В орбитальный базис были включены маффин-тин орбитали, соответствующие 7s, 6p, 6d и 5f состояниям Pu, 4s, 4p и 3d состояниям Ni и 4s, 4p и 4d состояниям Ga. При расчетах использованы величины параметров прямого кулоновского 4 эВ и обменного хундовского 0,48 эВ взаимодействий [3].
Результаты исследования и их обсуждение
Для соединений данной серии, так же как и многих других соединений 5f элементов, особенности электронной структуры связаны с наличием сильных кулоновских корреляций, сравнимых по величине с шириной 5f зоны. При этом сильное спин-орбитальное взаимодействие (СОВ) имеет величину порядка обменного взаимодействия, поэтому магнитное состояние определяется конкуренцией этих двух взаимодействий. Для учета всех перечисленных типов взаимодействий в исследовании применялся зонный метод LDA + U + SO, в котором одновременно учитываются кулоновское взаимодействие и СОВ [3, 4]. В данной работе представлены результаты исследования электронной структуры и магнитного состояния соединений PuCoGa5 и PuNiGa5 под давлением в модели всестороннего сжатия ячейки в рамках LDA + U + SO. В расчетах спиновый момент для ионов Pu в PuNiGa5 при приложении давления уменьшался с 1,17 [4] при нормальном давлении до 0,6 в случае объема, равного 0,58, от равновесного объема ячейки при нормальном давлении, что соответствовало падению полного момента в два раза с 0,22 до 0,11. Эффективный момент ионов Pu в PuNiGa5, вычисленный из закона Кюри – Вейсса, можно оценить, исходя из промежуточного типа связи как 0,47 магнетона Бора при нормальном давлении [4] и 0,25 магнетона Бора при 0,58 равновесного объема. Таким образом, для рассмотренного нами диапазона объемов элементарной ячейки PuNiGa5 величина эффективного магнитного момента ионов Pu в PuNiGa5, оцененная из закона Кюри – Вейсса и промежуточного типа связи уменьшается практически в 2 раза.
Полная и парциальные плотности состояний N для соединения PuNiGa5 приведены на рис. 1. Рассчитанные для соединения PuNiGa5 в рамках метода LDA + U + SO кривые приведены для значений объема элементарной ячейки 0,96 V0 – объема при нормальном давлении (верхний рисунок), 0,82 V0 (центральный рисунок) и 0,58 V0 (нижний рисунок). Уровень Ферми соответствует нулю на шкале энергий E (эВ). Из приведенных рисунков видно, что электронная структура 5f состояний Pu расщепилась на две подзоны – заполненную ниже уровня Ферми, с величиной полного момента 5/2, и подзону с 7/2, заполненную незначительно. Проведенный нами анализ матриц заселенности 5f оболочки ионов Pu показал, что электронная конфигурация всех исследованных соединений близка к f6, но тип связи близок к промежуточному.
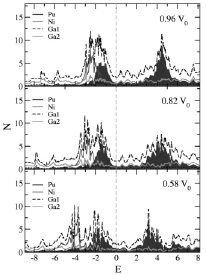
Рис. 1. Полная (темная пунктирная кривая) и парциальные Pu (серые области от –2 до 0 эВ и до 4 эВ выше уровня Ферми), Ni (серая кривая), Ga1 (мелкий пунктир) Ga2 (светлая кривая) плотности электронных состояний N (состояний/эВ на формульную единицу), рассчитанные для соединения PuNiGa5 в рамках метода LDA + U + SO для значений объема элементарной ячейки 0,96 V0 – объема при нормальном давлении (верхний рисунок), 0,82 V0 (центральный рисунок) и 0,58 V0 (нижний рисунок). Уровень Ферми соответствует нулю на шкале энергий E (эВ)
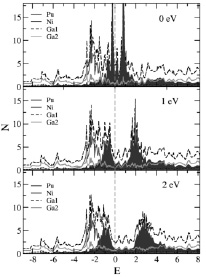
Рис. 2. Полная (темная пунктирная кривая) и парциальные Pu (серые области от –2 до 0 эВ и до 4 эВ выше уровня Ферми), Ni (серая кривая), Ga1 (мелкий пунктир) Ga2 (светлая кривая) плотности электронных состояний N (состояний/эВ на формульную единицу), рассчитанные для соединения PuNiGa5 в рамках метода LDA + U + SO для значений параметра U, равного 0 эВ (верхний рисунок), 1 эВ (центральный рисунок) и 2 эВ (нижний рисунок). Уровень Ферми соответствует нулю на шкале энергий E (эВ)
На верхнем рисунке полная (темная пунктирная кривая) и парциальные Pu плотности состояний, которые показаны залитой серой областью от –2 до 0 эВ и широким пиком с центром в районе 4 эВ выше уровня Ферми, дают основной вклад в плотность состояний. Серая кривая с основной плотностью состояний от –1 до –4 эВ соответствует электронным состояниям Ni. Видно, что для центрального и нижнего рисункам, соответствующим случаям сильного всестороннего сжатия ячейки PuNiGa5 происходит увеличение ширины Pu, Ni и других электронных состояний до 5 эВ в случае заполненной и пустой частей 5f состояний Pu в объеме 0,58 равновесного объема при нормальных условиях.
На рис. 2 приведены полученные результаты исследования влияния силы электронных корреляций на электронную структуру исследованного соединения PuNiGa5 – для этого проведены расчеты для серии значений (0, 1, 2 эВ) параметра кулоновского взаимодействия U в 5f оболочке Pu. Полная (темная пунктирная кривая) и парциальные Pu (серые области от –2 до 0 эВ и до 4 эВ выше уровня Ферми), Ni (серая кривая), Ga1 (мелкий пунктир) Ga2 (светлая кривая) – плотности электронных состояний N.
При повышении значения данного параметра увеличивается расстояние между пустыми и заполненными электронными состояниями Pu с 1 эВ для значения параметра U, равного 0 эВ (верхний рисунок), до 4 эВ для значения параметра U, равного 2 эВ (нижний рисунок). Эта величина включает спин-орбитальное расщепление между уровнями и обменное взаимодействие. Уровень Ферми соответствует нулю на шкале энергий E (эВ). При этом состояния никеля и галлия практически не сдвигаются, располагаясь преимущественно от –4 до 0 эВ в случае никеля и по всему энергетическому интервалу в случае Ga2.
Заключение
При помощи первопринципных самосогласованных расчетов исследованы изменения электронной структуры и магнитного состояния интерметаллида PuNiGa5 при всестороннем сжатии элементарной ячейки. Проведены расчеты электронной структуры данного интерметаллида в рамках первопринципного метода LDA + U + SO с учетом сильных электронных корреляций в 5f оболочке ионов плутония. Проведенный анализ электронной структуры показал, что при уменьшении объема ячейки интерметаллида PuNiGa5 вследствие всестороннего сжатия происходит увеличение ширины зон электронных состояний. Эффективный момент ионов Pu в PuNiGa5, вычисленный из закона Кюри – Вейсса, был оценен, исходя из промежуточного типа связи. При всестороннем сжатии элементарной ячейки в PuNiGa5 эта величина значительно уменьшается. Также проведены расчеты для исследования влияния силы электронных корреляций на электронную структуру соединения PuNiGa5. Для чего были проведены расчеты для серии значений (0, 1, 2 эВ) параметра кулоновского взаимодействия в 5f оболочке актиноидного металла, которые показали постепенное увеличение расстояния между пустыми и заполненными электронными состояниями Pu, которое также определяется величинами спин-орбитального расщепления и обменного взаимодействия данных электронных состояний.
Работа выполнена по проекту № 18-10-2-6 комплексной программы УрО РАН.