

Использование синтетических мРНК в исследовательской деятельности, а также производство таких молекул в промышленных масштабах при создании РНК-вакцин позволяет решать различные научные и терапевтические задачи, мРНК-вакцины оказались одним из наиболее эффективных инструментов в борьбе с недавней пандемией [1]. Производство этих препаратов зависит от получения высококачественной мРНК, синтезируемой посредством транскрипции in vitro [2]. Основным ферментом синтеза РНК in vitro является ДНК-зависимая РНК полимераза из бактериофага Т7 [3, 4]. Транскрипция РНК in vitro осуществляется со специально подготовленной ДНК матрицы, которая перед целевой последовательностью, содержащей открытую рамку считывания, а также 5’ и 3’ некодирующие области, имеет нуклеотидную последовательность Т7 промотора. Для получения трансляционно-активной мРНК in vitro, в нее необходимо внести две основные модификации после транскрипции. Первой модификацией является реакция синтеза 5’-концевого динуклеотида, называемой кэпированием, которая осуществляется в процессе транскрипции или после него [5, 6]. Вторая модификация, определяющая стабильность мРНК и степень ее трансляции в клетках, это достраивание поли(А) хвоста молекулы РНК с 3’ конца [7, 8].
В природе мРНК, синтезируемая в ядре, подвергается различным модификациям, прежде чем она может транслироваться в белки цитоплазмы. Для того чтобы мРНК была функциональной, она требует модифицированных 5´ и 3´ концов, а также кодирующей области (то есть открытой рамки считывания (ORF), кодирующей интересующий белок), фланкированной нетранслируемыми областями (5’ UTR и 3’UTR). Транскрибируемая мРНК (пре-мРНК) подвергается двум значительным модификациям в дополнение к сплайсингу. Во время синтеза структура 7-метилгуанилата, также известная как «кэп», добавляется к 5´ концу пре-мРНК через 5´ → 5´ трифосфатную связь. Этот кэп защищает зрелую мРНК от деградации, а также играет роль в ядерном экспорте и эффективной трансляции [9, 10]. Вторая модификация происходит посттранскрипционно на 3´ конце формирующейся молекулы РНК и характеризуется добавлением примерно 200 адениновых нуклеотидов – поли(A) хвоста. Эта модификация придает стабильность мРНК, способствует экспорту мРНК в цитозоль и участвует в образовании трансляционно-компетентного рибонуклеопротеина вместе со структурой 5´ кэпа. Зрелая мРНК образует кольцевую структуру (замкнутую петлю), соединяя кэп с поли(А) хвостом через кэп-связывающий белок eIF4E (эукариотический фактор инициации 4E) и поли(А)-связывающий белок PABP, оба из которых взаимодействуют с eIF4G (фактор инициации эукариот 4G) [10, 11]. Таким образом, основные требования к функциональной мРНК – это наличие 7-метилгуанилатного кэпа на 5´ конце и поли(A) хвоста на 3´ конце. Поэтому эти структуры должны быть добавлены после реакции транскрипции in vitro для получения эффективной трансляции синтетических молекул мРНК в эукариотических клетках.
Стратегии синтеза мРНК in vitro варьируются в зависимости от желаемого масштаба синтеза. Синтетическая РНК может быть эффективно синтезирована in vitro в реакции транскрипции с помощью прокариотических фаговых полимераз. Кэп структура и поли(А) хвост, характерные для зрелой мРНК, могут быть добавлены либо во время синтеза РНК или уже после реакции транскрипции при помощи отдельных ферментативных реакций с кэпирующими ферментами и поли(А)-полимеразой соответственно.
Реакции получения функциональных РНК in vitro хорошо масштабируются и могут осуществляться с использованием комплексного набора отдельных реагентов, в том числе ряда ферментов для синтеза и модификации РНК.
Целью настоящей работы являлась разработка технологии получения активных препаратов ферментов посттранскрипционной модификации молекул РНК, полученных in vitro.
Материалы и методы исследования
Получение генетических конструкций
Нуклеотидные последовательности, кодирующие поли(А)-полимеразу из E. coli, а также кэпирующие белки из вируса осповакцины синтезировали de novo из олигонуклеотидов. Полученные фрагменты ДНК амплифицировали ПЦР с использованием высокоточной ДНК-полимеразы Q5 (NEB) и олигонуклеотидов (таблица), содержащих уникальные сайты рестрикции NcoI и NotI для последовательностей, кодирующих поли(А)-полимеразу из E. coli и 2-О-метилтрансферазу вируса осповакцины либо XbaI и XhoI для генов, кодирующих субъединицы D1 и D12 кэпирующего фермента из вируса осповакцины. Для аффинной очистки ферментов использовали полигистидиновую метку (6His), кодируемую на N-конце аминокислотных последовательностей. Фрагменты ДНК чистили при помощи агарозного геля набором для выделения из агарозы (Евроген). ДНК-фрагменты после очистки использовались в реакции рестрикции с соответствующими эндонуклеазами рестрикции (37 °С, 1 ч). Плазмиды pET28a и pET-Duet использовали в реакции рестрикции этими же эндонуклеазами рестрикции: NdeI и NotI (для pET28a), а также XbaI и XhoI (для pET-Duet), а далее чистили резаный вектор через агарозу. Реакцию лигирования проводили при 22 °С в течение 1 ч при помощи ДНК-лигазы Т4, а потом инактивировали ДНК-лигазу прогревом при 65 °С в течение 10 мин и трансформировали полученной смесью компетентные клетки E. cloni 10G (Lucigen). Полученную бактериальную культуру высевали на чашки Петри с агаризованной средой LB, содержащими селективный антибиотик (ампициллин) и инкубировали в течение 12 ч при 37 °С до появления единичных колоний. Из полученных колоний (по 8 шт. для каждой генетической конструкции) выделяли плазмидную ДНК и секвенировали.
Олигонуклеотиды для клонирования генов, кодирующих белки модификации РНК в экспрессионные векторы pET28a и pET-Duet
|
polyA_Nco_f |
GATATACCATGGGAcatcatcaccaccatcacTTTACCCGAGTCGCTAATTTTTGC |
|
polyA_Not_r |
CTCGAGTGCGGCCGCtcaTGCGGTACCCTCACGACGTGGTG |
|
2Ometh_Nco_f |
GATATACCATGGGAGATGTTGTTTCTCTGGACAAACC |
|
2Ometh_Not_r |
CTCGAGTGCGGCCGCtcaGTGGTGATGATGGTGGTGTTCCAGTG |
|
D1D12_Xba_f |
GATATATCTAGAAATAATTTTGATTTAACTTTAAGAAG |
|
D1D12_Xho_r |
ctttaccagactcgagTTACAGCAGCAGTTTCACCAG |
Наработка белков для модификации синтетических мРНК в клетках E. Coli
Плазмидами с корректными вставками, содержащими гены белков для модификации РНК, трансформировали один из экспрессионных штаммов E. coli, предназначенных для продукции рекомбинантных белков. Трансформационную смесь высевали на чашки Петри с агаризованной средой и селективным антибиотиком (ампициллин, 100 мкг/мкл) и инкубировали ночь при 37 °С до появления единичных колоний. Затем единичной колонией инокулировали 5 мл жидкой питательной среды LB, содержащей ампициллин, и растили ночь при 37 °С на шейкере (Eppendorf Innova, 180 об/мин). Наработка рекомбинантных белков проводилась в культуре бактерий E. coli на среде LB (10 г/л триптон, 5 г/л дрожжевой экстракт, 5 г/л хлорид натрия). Ночной культурой инокулировали жидкую питательную среду LB в качалочных колбах (1/10 объема) и растили при 37 °С до оптической плотности OD600 = 0,6 единиц. Далее, для индукции экспрессии гена ДНК-зависимой РНК-полимеразы Т7 в среду добавляли ИПТГ (0,1 М) и инкубировали клеточную культуру при 37 °С 2–3 ч. Биомассу собирали центрифугированием при 5000 g в течение 30 мин при 4 °С.
Хроматографическая очистка белков для модификации синтетических мРНК
Клеточный осадок ресуспендировали в буфере А (50 мМ Трис-HCl pH 7,5; 0,3 М NaCl; 0,005 М имидазола) на льду. Далее бактериальные клетки разрушали ультразвуком во льду до просветления суспензии. Полученный лизат центрифугировали (10000 g, 40 мин при 4 °С) до осаждения клеточного дебриса и визуального просветления супернатанта. Надосадочную жидкость после ультразвукового разрушения биомассы и центрифугирования клеточного дебриса фильтровали через мембрану с размером пор 0,22 мкм. Полученный супернатант наносили на колонку с сорбентом Ni-NTA (Qiagen), предварительно промытую буфером (50 мM калий-фосфатного буфера рН 7,6, 300 mМ NaCl, 5 мM имидазола). Связавшийся с сорбентом белок элюировали хроматографическим буфером В (50 мМ Трис-HCl pH 7,5; 0,5 М имидазола). Фракции, содержащие максимальный уровень белка, объединяли и диализовали против буфера для хранения (40 мM HEPES-KOH pH 7,6; 100 мM KCl) с глицерином на -20 °С. Для избежания деградации РНК рибонуклеазами все растворы готовились на воде, обработанной DEPC. Электрофорез в полиакриламидном геле (ПААГ) проводили по стандартной методике Лэммли. Для белкового электрофореза в денатурирующих условиях использовался 8–12 % ПААГ. К белковому препарату добавлялся двукратный объем буфера для нанесения (250 мМ Трис-HCl pH 6,8; 6 % SDS, 2 % меркаптоэтанол, 16 % глицерин, 0,05 % бромфеноловый синий), раствор тщательно перемешивали и выдерживали в кипящей водяной бане в течение 5 мин. Окраску геля проводили с использованием Кумасси бриллиантового синего R-250. Для электрофореза использовали в трис-глициновый буфер (10х 1 % SDS, 0,25 М Трис-OH и 1,8 М глицин) на приборе фирмы Bio-Rad (CША).
Результаты исследования и их обсуждение
Эукариотические мРНК представляют собой сложные молекулы, состоящие из различных нуклеотидных участков, выполняющих свою функцию в процессе трансляции и стабильности РНК в клетках. Синтетические мРНК, которые получены in vitro, также должны подвергаться двум основным посттранскрипционным модификациям для трансляционной активности в клетках эукариот. Существует два варианта получения функционально-активных искусственных мРНК в реакции транскрипции in vitro в зависимости от выбранной стратегии кэпирования: а) стандартный синтез с последующим кэпированием при помощи специальных ферментов после реакции транскрипции (посттранскрипционное кэпирование); б) включение аналога кэпа во время транскрипции (ко-транскрипционное кэпирование). Выбор метода будет зависеть от масштаба необходимого синтеза мРНК и количества транскрибируемых матриц ДНК. Если предполагается стратегия с посттранскрипционной модификацией, то для реакции кэпирования используются вирусные кэпирующие ферменты, например, из вируса осповакцины (Vaccinia virus). Кэпирование происходит в две последовательные ферментативные реакции, в ходе которых сначала образуется структура кэп-0, а потом кэп-1. Трансляционная активность РНК с кэп-1 выше, что предпочтительнее для конечного продукта. Второй модификацией синтетических мРНК является процесс полиаденилирования, что предполагает добавку поли(А) хвоста с 3’ конца, который может быть либо закодирован в матрице ДНК с использованием праймера для ПЦР с соответствующей последовательностью, либо он может быть добавлен посттраснкрипционно к уже синтезированной РНК на стадии ферментативной обработки поли(A)-полимеразой, например, из E. coli. При выборе такой стратегии длину добавленного хвоста можно регулировать титрованием количества поли(А)-полимеразы в реакции. Таким образом, для посттранскрипционной модификации искусственных мРНК, которая позволит им транслироваться в эукариотических клетках, необходимы белковые препараты кэпирующих ферментов и поли(А)-полимеразы.
Фрагменты ДНК, кодирующие гены, выбранных белков, были синтезированы de novo из олигонуклеотидов и клонированы в бактериальные экспрессионные векторы под контролем Т7 промотора с возможностью индукции экспрессии ИПТГ. Для генов, кодирующих поли(А)-полимеразу и 2-О-метилтрансферазу вируса осповакцины, был использован вектор pET28a, куда по сайтам рестрикции NcoI и NotI были вставлены соответствующие фрагменты ДНК. Карты полученных генетических конструкций приведены на рис. 1.
Фрагменты ДНК, соответствующие генам субъединиц основного кэпирующего фермента вируса осповакцины D1 и D12, были клонированы по сайтам XbaI и XhoI в вектор pET-Duet, который позволяет индуцируемо экспрессировать сразу два гена одновременно с отдельных промоторов Т7. Карта генетической конструкции, экспрессирующей оба гена кэпирующего фермента, приведена на рис. 2. После наработки в клетках обе субъединицы формируют двухсубъединичный фермент, который можно очистить при помощи аффинной хроматографии за полигистидиновый хвост (6His), который закодирован на N-конце большой субъединицы D1. Таким образом, количество наработанного кэпирующего фермента зависит от количества субъединицы D1, поскольку меньшая субъединица D12 нарабатывается с избытком по отношению к большой из-за различий в размере.
Общая схема получения штаммов-продуцентов была следующая. Полученными плазмидами трансформировали компетентные клетки штамма E. coli BL21(DE3), созданного для продукции рекомбинантных белков. После получения единичных бактериальных колоний, содержащих соответствующие плазмиды, проводили засев ночных культур в микробиологических пробирках для получения стартовых культур перед культивацией в больших объемах.
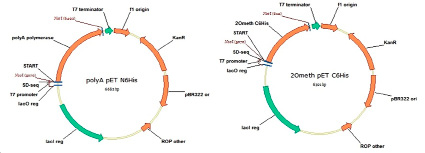
Рис. 1. Карта экспрессионных векторов для продукции фермента поли(А)-полимеразы из E. coli (левая панель) и фермента 2-О-метилтрансферазы вируса осповакцины под контролем промотора Т7 на основе коммерческого вектора pET28a
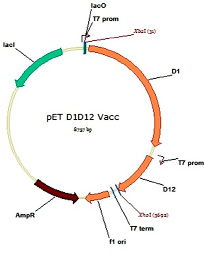
Рис. 2. Карта экспрессионного вектора для продукции основного кэпирующего фермента вируса осповакцины, состоящего из двух субъединиц (большой D1 и малой D12) под контролем двух Т7 промоторов для одновременной экспрессии обоих генов на базе вектора pET-Duet
Для получения подходящего количества белка при хроматографической очистке использовали объемы культур, выращенные в 100–200 мл жидкой среды LB с соответствующими антибиотиками (канамицин для вектора pET28a, ампициллин для вектора pET-Duet). После подроста бактериальных культур и индукции при помощи ИПТГ собирали биомассу и разрушали клетки, как описано в разделе «Материалы и методы исследования». После осаждения клеточного дебриса отфильтрованный через фильтр 0,22 мкм надосадок наносили на хроматографическую колонку, содержащую метал-хелатный сорбент Ni-NTA (Qiagen).
Так, детальнее процесс получения фермента поли(А)-полимераза из E. coli, для очистки которого подбирали условия, выглядел следующим образом. Штамм E. coli BL21(DE3), содержащий плазмиду с клонированным геном, кодирующим поли(А)-полимеразу, культивировали в объеме жидкой культуры LB 100 мл. Культивирование производили при 37 °С на шейкере при 150 об/мин. Оптическая плотность при внесении индуктора ИПТГ составляла OD = 0,6 единиц. Оптическая плотность при сборе биомассы была OD = 1,2 единиц (3 ч после индукции). Биомассу собирали центрифугированием при 5000 g в течение 30 мин при 4 °С. Далее ресуспендировали клетки в 5 мл буфера А (50 мМ Трис-HCl pH 7,5; 0,3 М NaCl; 0,005 М имидазола). Разрушение биомассы осуществляли при помощи ультразвукового дезинтегратора на льду до просветления лизата (20–30 мин с интервалами между озвучиванием, в режиме 30 с УЗ, 5 мин пауза). Осаждали клеточный дебрис центрифугированием при 10000 g в течение 40 мин при 4 °С. Хроматографическую очистку осуществляли на аффинном сорбенте Ni-NTA, белок элюировали градиентом имидазола от 0 до 250 мМ. Анализировали экспрессию и очистку при помощи ПААГ электрофореза (рис. 3). После анализа объединяли фракции, содержащие целевой белок, и переводили в буфер для хранения при помощи диализа. Размер белка – 54кДа.
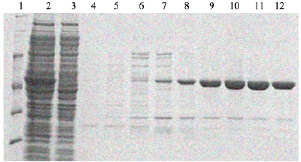
Рис. 3. Наработка и очистка поли(А)-полимеразы E. coli в штамме E. coli BL21(DE3) (1 – маркер, 2 – супернатант после разрушения клеток, наносимый на колонку, 3 – тельца включения, осадок растворен в 8 М мочевине, 4 и 5 – проскок и промывка хроматографической колонки, 6–12 – фракции после градиента элюции имидазолом от 5 до 250 мМ)
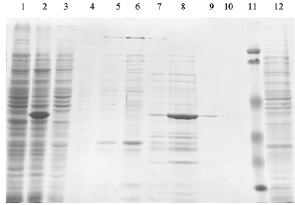
Рис. 4. Наработка и очистка 2-О-метилтрансферазы вируса осповакцины в штамме E. coli KRX (1 – клетки до индукции рамнозой, 2 – супернатант после разрушения клеток, наносимый на колонку, 3 и 4 – проскок и промывка хроматографической колонки, 5–10 – фракции после градиента элюции имидазолом от 10 до 250 мМ, 11 – маркер, 12 – тельца включения, осадок растворен в 8 М мочевине)
Для получения чистого препарата 2-О-метилтрансферазы вируса осповакцины были подобраны следующие компоненты и условия. Штамм KRX (Promega) трансформированный генетической конструкцией, которая несет в себе требуемый ген, культивировали в жидкой культуре LB в объеме 100 мл при температуре 37 °С. Оптическая плотность при внесении индуктора рамнозы была OD = 0,4 единиц. Далее, продолжали инкубацию бактериальной культуры до оптической плотности OD = 1 единиц (3 ч после индукции). Собирали биомассу как описано выше и ресуспендировали в 4 мл буфера для разрушения (Трис, 8, 50 мМ, NaCl, 500 mM). Разрушение биомассы и подготовку осветленного лизата для нанесения на хроматографическую колонку осуществляли аналогично описанному для поли(А)-полимеразы: разрушение при помощи УЗ на льду до просветления лизата (20–30 мин с интервалами между озвучиванием); осаждение клеточного дебриса: центрифугирование при 10000 g в течение 40 мин. Далее осуществляли хроматографическую очистку на металл-хелатном сорбенте NiNTA, а элюцию связавшегося белка проводили имидазолом с градиентом концентрации 0–250 мМ. Перевод в буфер для хранения: диализ. Размер белка – 40 кДа. Анализ наработки и очистки представлен на рис. 4.
Основной белок для кэпирования из вируса осповакцины состоит из двух субъединиц: большой D1 и малой D12. Для одновременной наработки обеих субъединиц был использован специальный экспрессионный вектор pET-Duet, позволяющий одновременную индукцию экспрессии обоих генов, кодирующих субъединицы фермента. При наработке обеих субъединиц они формируют единый ферментативный комплекс, аффинную очистку которого можно осуществить за полигистидиновый хвост, кодируемого на большой субъединице D1. Поскольку при наработке в штамме E. coli BL21 (DE3) практически весь целевой белок переходил в форму нефункциональных агрегатов, то в качестве подходящего штамма для наработки этого фермента был выбран штамм E. coli T7 ExpressLysY со стадией инкубации при пониженных температурах (16 °С). Детально протокол наработки и очистки выглядел следующим образом. После трансформации штамма E. coli T7 ExpressLysY плазмидой, кодирующей обе субъединицы кэпирующего фермента, клеточную культуру растили до оптической плотности OD = 0,5 единиц. Затем бактериальную культуру охлаждали на льду не менее 5 мин и добавляли до 0,1 мМ IPTG, после чего следовала инкубация при интенсивном перемешивании на 16 °С в течение 12 ч. Клеточную культуру после инкубации охлаждали на льду и осаждали клетки центрифугированием, как указано в разделе «Материалы и методы исследования».
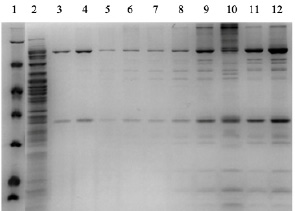
Рис. 5. Наработка и очистка обеих субъединиц (большой D1 и малой D12) основного кэпирующего фермента вируса осповакцины в штамме E. coli T7 ExpressLysY (1 – маркер, 2 – супернатант после разрушения клеток, наносимый на колонку, 3–10 – фракции после градиента элюции имидазолом от 10 до 250 мМ, 11 и 12 – объединенные и сконцентрированные хроматографические фракции, содержащие целевой белковый продукт)
После осаждения клеток ресуспендировали осадки в 1/10 от объема клеточной культуры в буфере (50 mM TrisHCl pH8, 300 mM NaCl, 5 mM Imidazole, 5 mM DTT). Клетки разрушали с помощью ультразвукового дезинтегратора, а полученный препарат центрифугировали и фильтровали, как описано ранее.
Полученный клеточный лизат смешиваем с 0,5 мл IMAC, инкубация с качением в течение 1 ч. Промываем сорбент 50 mM TrisHCl pH8, 300 mM NaCl, 5 mM Imidazole, 0,1 % Triton x-100, 5 mM DTT 10V колонки. Элюируем 50 mM TrisHCl pH8, 300 mM NaCl, 500 mM Imidazole, 10 % глицерол, 5 mM DTT, снимаем 4 фракции по 1 мл. В белковые фракции после элюции добавляется Triton x-100 до 0,2 %, далее проверяем с помощью э/фореза наличие целевого белка и проводим диализ (все фракции раздельно) против 40 mM TrisHCl pH8, 200 mM NaCl, 0,2 mM ЭДТА, 10 % глицерол, 5 mM DTT (буфер дополнительно фильтровали в центриконах на 3 кДа), затем фракции фильтруем через 0,2 um фильтр. После проверки на РНКазную активность фракцию 1 сконцентрировали и довели содержание глицерола до 50 %. Анализ наработки и очистки представлен на рис. 5.
Таким образом, в ходе настоящей работы были получены штаммы-продуценты и подобраны условия для наработки трех рекомбинантных белков – ферментов, необходимых для посттранскрипционной модификации синтетических мРНК. Фермент поли(А)-полимераза необходим для модификации 3’ конца молекулы РНК для достраивания поли(А) хвоста, а оба кэпирующих фермента из вируса осповакцины позволяют осуществить посттранскрипционное кэпирование РНК, что необходимо для формирования трансляционного комплекса и стабильности мРНК в клетке [12, 13].
Заключение
В данной работе были подобраны условия для получения ферментов модификации синтетических мРНК – поли(А) полимеразы из E. coli, а также двух кэпирующих ферментов из вируса осповакцины. В ходе экспериментов были найдены оптимальные условия для продукции и очистки этих ферментов. Полученные препараты могут быть использованы для посттранскрипционной модификации синтетических РНК in vitro и получения функционально-активных молекул мРНК для исследовательских и терапевтических целей.
Настоящая статья не содержит каких-либо исследований с участием людей или животных в качестве объектов исследований.
Авторы заявляют об отсутствии конфликта интересов.